- 4
- May
- 0


OSTEOGENESIS IMPERFECTA
OSTEOGENESIS IMPERFECTA
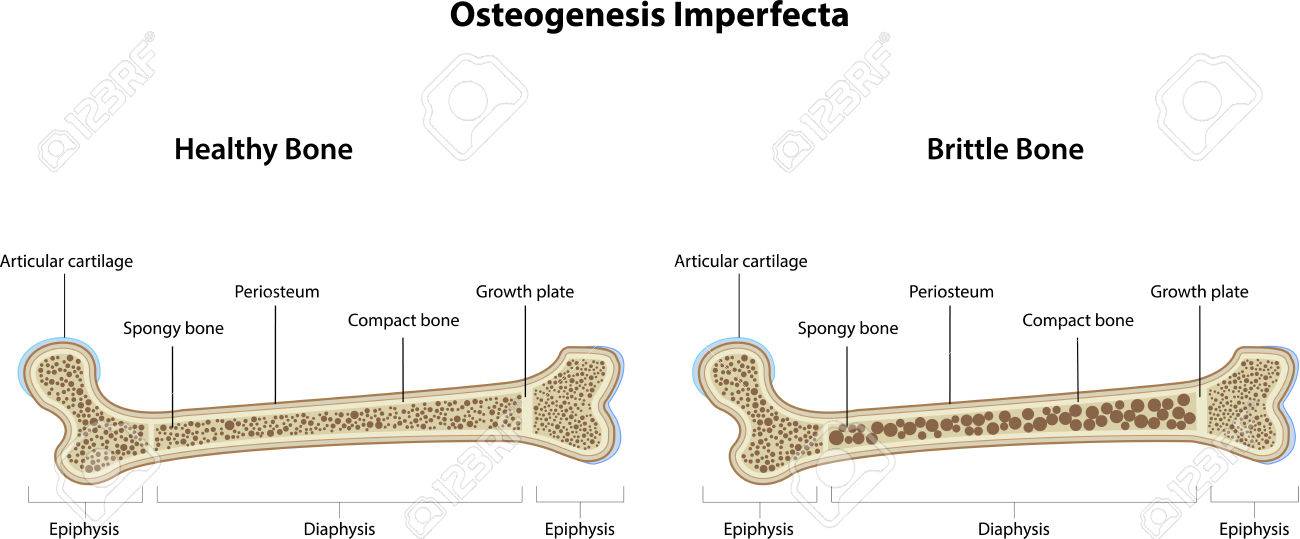
En medicina, la osteogénesis imperfecta u osteogenia imperfecta (también llamada huesos de cristal) es un trastorno congénito, es decir,
presente al nacer, que se caracteriza por una fragilidad de hueso excesiva, como consecuencia de una deficiencia congénita en la elaboración
de una proteína, el colágeno tipo I. Quienes portan el defecto tienen menos colágeno de lo normal o es de una menor calidad y como es una
proteínaimportante en la estructura de los huesos, causa una fragilidad y debilidad poco usual de los huesos.1 El diagnóstico es radiológico,
incluso antes del parto.
Clasificación de Sillence
- Tipo 1: Enfermedad de Ekman Lobstein, autosómica dominante. Se caracteriza por osteoporosis generalizada con fragilidad ósea anormal,
escleróticas azules toda la vida, hay una reducción del espesor esclerar y la uvea pigmentada subyacente se hace visible a través de la
eclerótica adelgazada, pérdida de la audición de tipo conductivo presenil, la que puede ser de conducción por otoesclerosis o tipo nervioso
por la compresión del nervio VIII al salir del cráneo y la otoesclerosis es resultado de la proliferación anormal del cartílago, el cual al
calcificarse produce esclerosis de la porción petrosa del temporal. No todos los pacientes presentan dentinogénesis imperfecta. COL1A1.  Tipo 2: Enfermedad de Vrolik, autosómica recesiva. Se caracteriza por fragilidad ósea extraordinaria, culmina en la muerte en el periodo perinatal o comienzos de la lactancia, huesos largos muy fragmentados (fémur en acordeón), prominencia de hueso parietal y temporal con occipucio colgante y osificación de cráneo retrasada extraordinariamente. COL1A1 ó COL1A2.
Tipo 2: Enfermedad de Vrolik, autosómica recesiva. Se caracteriza por fragilidad ósea extraordinaria, culmina en la muerte en el periodo perinatal o comienzos de la lactancia, huesos largos muy fragmentados (fémur en acordeón), prominencia de hueso parietal y temporal con occipucio colgante y osificación de cráneo retrasada extraordinariamente. COL1A1 ó COL1A2.
- Tipo 3: Autosómica recesiva. Caracterizada por fragilidad ósea intensa ocasionando múltiples fracturas, deformidad progresiva de huesos
largos, retardo grave del crecimiento con la talla más pequeña de todas las OI, escleróticas azules en el neonato pero con la edad se vuelven
menos azules, Dentinogénesis imperfecta, deformidad de columna por combinación de osteoporosis intensa, fracturas por compresión de vértebras
e hiperlaxitud ligamentosa, cifoescoleosis, siendo la más común la escoliosis dorsal, cara aspecto triangular de “duende”, frente amplia, prominencia
de huesos parietal, temporal y con occipucio colgante. COL1A1 ó COL1A2. - Tipo 4: Autosómica dominante. En el neonato las escleras tienen color normal, pueden llegar a estar azulados, sin embargo, se vuelven cada vez menos
azules, osteoporosis, fragilidad ósea y deformidad de huesos largos, si presenta dentinogénesis imperfecta se clasifica en la subdivisión 4 A, si no presenta
dentinogénesis imperfecta se clasifica 4 B. COL1A1 ó COL1A2. En la dentinogésis imperfecta hay alteraciones por deficiencia de dentina, manchas pardo
amarillentas o azulosas grisáceas translúcidas y los incisivos inferiores son los más afectados. - Tipo 5: Autosómica dominante. Es similar a la tipo 4. Es más común que se presente en niños que muestran escleras blancas, formación de callo hiperplásico,
más común en fémur, tibia y húmero. En radiografías se advierte la formación masiva de callos en forma de mariposa, calcificación de la membrana interósea
en el antebrazo y en consecuencia problemas en la pronosupinación de miembros superiores, dislocación de la cabeza radial anterior. IFITM5. - Tipo 6: Autosómica recesiva. Fenotipo de moderado a severo, se presentan fracturas en los dos primeros años de vida, escleróticas normales o azul claro
, fracturas de vértebras, en esta tipo no hay dentinogenesis imperfecta. SERPINF1. - Tipo 7: Autosómica recesiva. Fenotipo perinatal letal o severo no-letal, rizomelica, acortamiento de caderas y hombros y un significativo encorvamiento. CRTAP.
- Tipo 8: Autosómica recesiva. Fenotipo perinatal letal o severo no-letal, huesos largos poco mineralizados y epífisis bulbosa. LEPRE1
Tratamiento
Un tratamiento integral de la osteogénesis imperfecta debe sostenerse fundamentalmente en 3 pilares: tratamiento farmacológico, ortopédico y rehabilitador y
acompañarse además, según las necesidades del paciente, de seguimiento por otros especialistas como el odontólogo, psicólogo, reumatólogo, endocrinólogo, cardiólogo, otorrinolaringólogo, etc.
Leave a Comment